
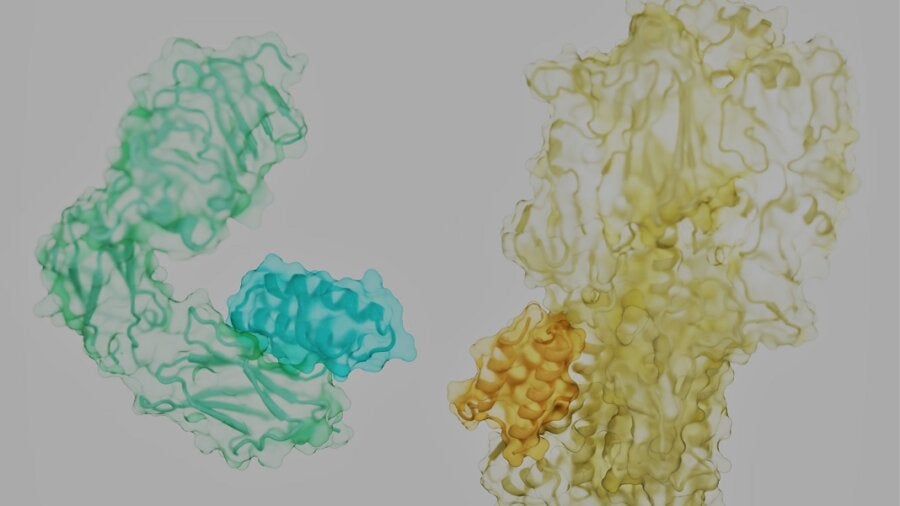
The proteins that control our lives are like rolling tumbleweeds. Each has a tangled, unique shape, with spiky side-branches dotting its surface. Hidden in the nooks and crannies are the locks to battle our most notorious foes—cancer, diabetes, infections, or even aging—if we can find the right key.
We just got a universal key maker. In a preprint, a team led by Dr. David Baker from the University of Washington developed an algorithm to design tiny protein keys that unlock those targets from scratch. Far from an ivory tower pursuit, the algorithm tackled one of the most head-scratching drug discovery challenges of our times: can we design drugs based on the structure of a protein’s lock alone?
They’re not talking about just any drug. Rather than focusing on small molecules, such as Tylenol, the team turned their attention to protein-like molecules, dubbed “binders.” While they may sound exotic, you know them. Monoclonal antibodies are one example, which have been key for treating severe Covid-19 cases. They’re also some of our best weapons against cancer. But these therapeutic giants struggle to tunnel into cells, are difficult to manufacture, and are often prohibitively expensive for widespread use.
What about an alternative? Can we tap into the power of modern computation and design similar but smaller and simpler drugs that are just as—if not more—effective?
Based on the Baker team’s study, the answer is yes. Screening nearly half a million candidate binder structures for 12 protein targets, the algorithm aced its task, using minimal computational power compared to previous attempts and highlighting potential hits. It also found a “cheat code” that made binders more efficient at grabbing onto their targets.
Here’s the kicker: unlike previous tools, the software only needed the structure of the target protein to engineer binder “keys” from scratch. It’s a far simpler approach compared to previous attempts. And because proteins run our internal biological universe, it means the new software key makers can help us unlock the secrets of our cells’ molecular lives—and intervene when they go awry.
“The ability to generate new proteins that bind tightly and specifically to any molecular target that you want is a paradigm shift in drug development and molecular biology more broadly,” said Baker.
Protein Binder What?
Our bodies are governed by a vast consortium of proteins. Like courtesans in a ballroom, each protein bounces around the cell, temporarily grabbing onto another protein before leaving them to find the next. Specific pairings can launch cellular plots to trigger—or inhibit—dramatic cellular processes. Some may direct a cell to grow or to peacefully pass away. Others may turn a cell cancerous or senescent, leaking toxic chemicals and endangering nearby cells.
In other words, protein pairings are essential for life. They’re also a powerful hack for medicine: if any pair triggers a signaling cascade that injures a cell or tissue, we can engineer a “doorstop” molecule to literally break up the pairing and stop the disease.
The problem? Imagine trying to separate two intertwined tumbleweeds rolling down a highway by throwing a short but flexible stick at them. It seems an impossible task. But the new study laid out a recipe for success: the key is finding where to pry the two apart.
Up the Wall
Proteins are often described as beads on chains that are crumpled into sophisticated 3D structures. That’s not entirely correct. The molecular “beads” that make up proteins are more like humanoid robots, with a stiff trunk and floppy limbs called “side chains.”
As a protein assembles, it links the trunk components of its constituent amino acids into a solid backbone. Like a fuzzy ball of yarn, the frizz—exposed side chains—cover the protein’s surface. Depending on their position and the backbone, they form pockets that a natural protein partner, or a mimic, can readily grab onto.
Previous studies tapped into these pockets to design mimic binders. But the process is computationally hefty and often relies on known protein structures—a valuable resource not always available. Another approach is to hunt down “hot spots” on a target protein, but these aren’t always accessible to binders.
Here, the team took a stab at the problem in a way that’s analogous to rock climbers trying to scale a new wall. The climbers are the binders, the wall is the target protein surface. Looking up, there are plenty of handholds and footholds made of side chains and protein pockets. But the largest ones, the “hot spots,” can’t necessarily hold the climber for the route.
Another approach, the team explained, is to map out all the holds, even if some seem weak. This opens a new universe of potential binding spots—most will fail, but some combinations may surprisingly succeed. A subset of these points are then challenged with thousands of climbers, each trying to identify a promising route. Once the top routes emerge, a second round of climbers will explore these routes in detail.
“Following this analogy, we devised a multi-step approach to overcome” previous challenges, the team said.
To start, the team scanned a library of potential protein backbones and a massive set of sidechain positions that can latch onto a protein target.
The initial sample sizes were enormous. Thousands of potential protein backbone “trunks” and nearly one billion possible sidechain “arms” emerged for every target.
With the help of Rosetta, the protein structure and function mapping program that Baker’s team developed, the team narrowed down the selection to a handful of promising binders.
The selection of these binders relies on “traditional physics” without tapping into machine learning or deep learning powers, said Dr. Lance Stewart, chief strategy and operations officer at the Institute for Protein Design, where Baker’s lab is based. It “makes this breakthrough even more impressive.”
Guiding Life
The next big question: so the binders can bind in silico. But do they actually work in cells?
In a proof of concept, the team picked 12 proteins to test out their algorithm. Among these were proteins closely involved in cancer, insulin, and aging. Another group zoomed in on battling pathogens, including surface proteins on the flu or SARS-CoV-2, the virus behind Covid-19.
The team screened 15,000–100,000 binders for each of the protein targets, and tested top candidates in E. coli bacteria. As expected, the binders were highly efficient at blocking their targets. Some cut off growth signals that can lead to cancer. Others targeted a common region of influenza—the flu—that in theory could neutralize multiple strains, paving the way for a universal flu vaccine. Not even SARS-CoV-2 spared, with the “ultrapotent” binders providing protection against its invasion in mice (those results were previously published).
The study showed that it’s possible to design protein-like drugs from the ground up. All it takes is the structure of the target protein.
“The possibilities for application seem endless,” remarked Dr. Sjors Scheres, joint head of structural studies at the MRC Laboratory of Molecular Biology in Cambridge, UK, on Twitter, who wasn’t involved in the study.
The algorithm, though powerful, isn’t perfect. Despite finding millions of potential binders, only a small fraction of the designs actually latched onto their target. Even the best candidates needed multiple changes to their amino acid makeup for optimal binding to a target.
But it’s groundbreaking work for a field that could fundamentally change medicine. For now, the method and large dataset “provides a starting point” to figure out how proteins interact inside our cells. These data, in turn, could guide even better computational models in a virtuous circle, especially with an added dose of deep learning magic.
It’ll “further improve the speed and accuracy of design,” said Stewart. It’s “work that is already ongoing in our labs.”
Image Credit: Longxing Cao, Brian Coventry, David Baker, UW Medicine
* This article was originally published at Singularity Hub

0 Comments